Phylogeography of Equine Infectious Anemia Virus
Abstract
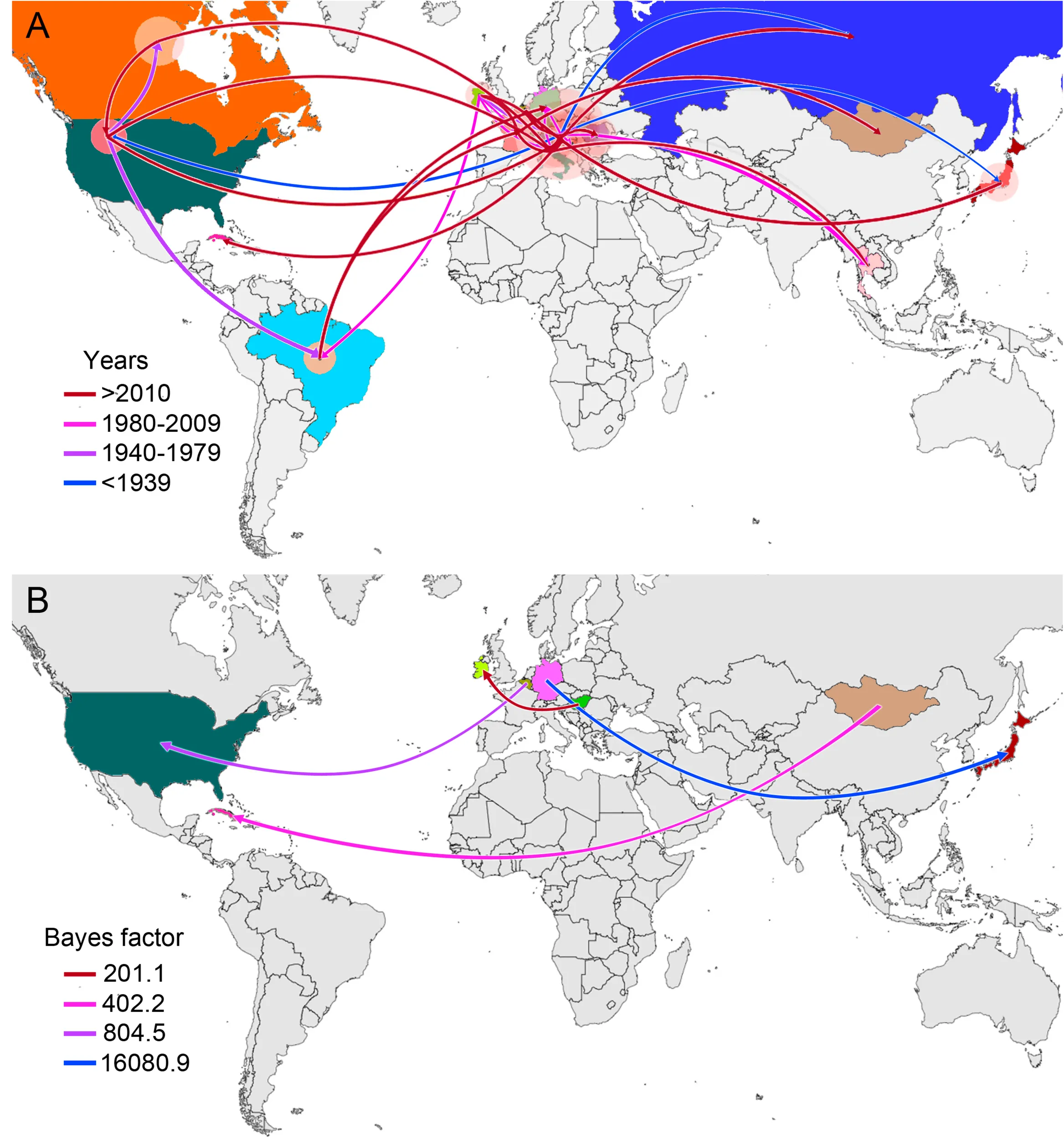
Equine Infectious Anemia virus (EIAV) is the causative agent of one of the most impacting infectious diseases affecting equids. EIAV is genetically diverse with several lineages circulating globally. To elucidate EIAV global spread patterns, we studied its spatiotemporal dynamics applying Bayesian phylodynamic analyses, using a worldwide compiled dataset composed of unique sequences of the gag gene. We also performed a scoping review of 1,470 publications on EIAV to characterize the spatiotemporal trends in EIAV research. Phylogeographic reconstruction suggested Hungary as the most likely country of origin for current EIAV circulation, and one of the most important centers of diversification. Historical EIAV spread was predominantly characterized by long-distance spread across continents. The American and Asian circulating EIAV lineages are more related to European lineages than to other Asian countries, with Europe being the continent with the highest EIAV phylogenetic diversity. Our bibliometric analysis showed a continuous increment in the number of publications per year, with the United States and China having the highest EIAV-related scientific production. This study provides a historical geographic mapping of EIAV lineage spread patterns and identifies important asymmetry between current research effort and availability of genetic data.